Directe octrooiinbreuk door de achter de woorden van de conclusies liggende uitvindingsgedachte
Hof Den Haag 16 februari 2016, LS&R 1265; ECLI:NL:GHDHA:2016:339 (Astrazeneca en Shionogi tegen Resolution)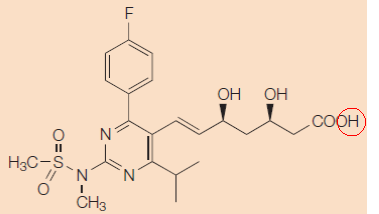
De achter de woorden van de conclusies liggende uitvindingsgedachte
5.3. Bij het achterhalen van de uitvindingsgedachte gaat het om de vaststelling van hetgeen het octrooi toevoegt aan de stand van de techniek en is het perspectief van de gemiddelde vakman en diens kennis van de stand van de techniek op de prioriteitsdatum richtinggevend (vgl. HR in Medinol / Abbott, r.o. 3.5.2). Daarnaast vormen de beschrijving en de tekeningen een belangrijke bron voor het achterhalen van de uitvindingsgedachte (vgl. HR in Medinol / Abbott, r.o. 3.4.2 hiervoor geciteerd). Niet in geschil is dat de gemiddelde vakman in dit geval een organisch chemicus is die zich bezig houdt met de ontwikkeling van nieuwe geneesmiddelen en die in elk geval een basiskennis heeft van de werking van de op de markt zijnde geneesmiddelen zoals rosuvastatine.
5.6. Gelet op de beschrijving in zijn geheel beschouwd, zal de gemiddelde vakman, in aanmerking genomen zijn algemene vakkennis op de prioriteitsdatum, begrijpen dat de uitvinding betrekking heeft op een nieuwe groep van statines – in het bijzonder het specifiek geclaimde rosuvastatine – waarvan de biologische activiteit beter is dan die van een bekende eerste generatie statines. De nieuwheid en inventiviteit van rosuvastatine is niet bestreden. Uitgaand van het perspectief van de gemiddelde vakman op de prioriteitsdatum wordt door het octrooi derhalve een nieuwe groep statines, waaronder meer specifiek rosuvastatine, aan de bekende stand van de techniek toegevoegd. Het vinden daarvan moet worden aangemerkt als de achter de woorden van de conclusie(s) liggende uitvindingsgedachte.
Uitleg van ‘of een niet-toxisch farmaceutisch aanvaardbaar zout daarvan’
5.22. Al het voorgaande leidt tot de conclusie dat de gemiddelde vakman op de prioriteitsdatum geen goede grond had om aan te nemen dat de octrooihouder zijn octrooi wilde beperken tot de in paragraaf 7 genoemde zouten voor gebruik met rosuvastatine en daarmee afstand wilde doen van een deel van de bescherming waarop hij ingevolge de letterlijke bewoordingen van conclusie 1 aanspraak kon maken. Of anders gezegd: de gemiddelde vakman zou niet aannemen dat de octrooihouder er bewust voor heeft gekozen alleen de in paragraaf 7 genoemde zouten van rosuvastatine onder bescherming te stellen. Hij zou de in paragraaf 7 gegeven opsomming daarom niet als limitatief beschouwen. Dat brengt met zich dat conclusie 1 aldus dient te worden uitgelegd dat de beschermingsomvang ervan zich (naast het rosuvastatinezuur) uitstrekt tot alle niet-toxische farmaceutisch aanvaardbare zouten van rosuvastatine, ook die niet zijn genoemd in paragraaf 7 van de beschrijving. Ook onafhankelijk van de ‘goede grond voor afstand-leer’ zou overigens op grond van hetgeen in r.o. 5.14 t/m 5.24 is overwogen tot dezelfde conclusie zijn gekomen.
Toegevoegde materie
5.31. Dat de gemiddelde vakman niet van te voren kon voorspellen of en in welke mate de zuurvorm en zoutvormen van rosuvastatine in de praktijk daadwerkelijk geschikt zouden blijken te zijn voor toepassing in een farmaceutisch preparaat met rosuvastatine, zoals Resolution stelt, staat aan de directe en ondubbelzinnige openbaarmaking in de aanvrage van de in conclusie 1 van EP 471 geclaimde zuurvorm en zoutvormen niet in de weg. Niet-werkende zoutvormen vallen niet onder conclusie 1 omdat daarin alleen farmaceutisch aanvaardbare zouten worden geclaimd. Overigens heeft Resolution, op wie terzake de stelplicht en zo nodig bewijslast rust, onvoldoende onderbouwd aangevoerd op grond waarvan moet worden aangenomen dat de gemiddelde vakman waterstof (waarmee het zuur wordt gevormd), of enig kation waarmee een zout van rosuvastatine gevormd kan worden, ondanks dat deze expliciet in de aanvrage worden genoemd, toch niet als reële mogelijkheid op positie R4 zou meelezen. Ten aanzien van de zuurvorm heeft Resolution gewezen op de publicatie van Berghe uit 1977 ((Pharmaceutical Salts, Journal of Pharmaceutical Sciences, Vol. 66, 1) waarin, niet specifiek ten aanzien van statines, is opgemerkt dat ‘most organic acids and bases are only poorly soluable in H2O’. Dit is evenwel onvoldoende, mede in het licht van de stelling van Astrazeneca, onder verwijzing naar verklaringen van haar partij-deskundigen [S] (3e verklaring par. 3-8 en de daarin genoemde publicatie van T.M. Serajuddin over verschillende statines (Journal of Pharmaceutical Sciences, Vol. 80, 9), waarin is geopenbaard dat polaire statines zoals rosuvastatine als vrij zuur redelijk goed oplosbaar zijn) en [F] (1e verklaring par. 6-11 ), dat de gemiddelde vakman het vrije zuur en een zout als vrijwel gelijk zal beschouwen. Dat conclusie 1 nawerkbaar is heeft Resolution ook niet bestreden.
5.32. Op grond van het voorgaande is het hof van oordeel dat geen sprake is van toegevoegde materie doordat conclusie 1 van EP 471 zich tevens uitstrekt tot rosuvastatinezuur en ook andere niet-toxische farmaceutisch aanvaardbare zouten dan het natrium- en calciumzout.
 ABC voor medische producten. Het hof verklaart voor recht:
ABC voor medische producten. Het hof verklaart voor recht: Octrooirecht. ABC. Afstandsleer. Shionogi is farmaceutische onderneming en houdster van ABC 300125 voor 'Rosuvastatinum, desgewenst in de vorm van een niet-toxisch farmaceutisch aanvaardbaar zout, in het bijzonder het calciumzout', voorheen houdster van EP471 'Pyrimidinederivaten als HMG-CoA-reductase-inhibitoren’. Resolution vordert met succes de partiële nietigheid van het octrooi en ABC vanwege toegevoegde materie; voor zover de bescherming ervan zich uitstrekt over andere producten dan in EP 471 genoemde niet-toxische farmaceutisch aanvaardbare zouten van rosuvastatine.
Octrooirecht. ABC. Afstandsleer. Shionogi is farmaceutische onderneming en houdster van ABC 300125 voor 'Rosuvastatinum, desgewenst in de vorm van een niet-toxisch farmaceutisch aanvaardbaar zout, in het bijzonder het calciumzout', voorheen houdster van EP471 'Pyrimidinederivaten als HMG-CoA-reductase-inhibitoren’. Resolution vordert met succes de partiële nietigheid van het octrooi en ABC vanwege toegevoegde materie; voor zover de bescherming ervan zich uitstrekt over andere producten dan in EP 471 genoemde niet-toxische farmaceutisch aanvaardbare zouten van rosuvastatine. Handelsvergunning. ABC. Diergeneesmiddel. Concept of eerste handelsvergunning in de EEA (European Economic Area). Antwoord:
Handelsvergunning. ABC. Diergeneesmiddel. Concept of eerste handelsvergunning in de EEA (European Economic Area). Antwoord: Octrooirecht. ABC. Vgl.
Octrooirecht. ABC. Vgl.  Uitspraak ingezonden door Martijn de Lange,
Uitspraak ingezonden door Martijn de Lange, ![image32193[1238].png](https://www.gtp.gr/MGFiles/logos/image32193%5B1238%5D.png) Conclusie AG:
Conclusie AG:  Octrooirecht. ABC. Begrip ‚werkzame stof’ – Pneumokokkenconjugaatvaccin – Pediatrisch gebruik – Dragerproteïne – Covalente binding. Het Hof beantwoordt de gestelde vragen [
Octrooirecht. ABC. Begrip ‚werkzame stof’ – Pneumokokkenconjugaatvaccin – Pediatrisch gebruik – Dragerproteïne – Covalente binding. Het Hof beantwoordt de gestelde vragen [













