EMA ongoing public consultations - update september 2013
![]() Hieronder een overzicht van de doorgaande publieke consultaties:
Hieronder een overzicht van de doorgaande publieke consultaties:
- European Medicines Agency, Concept paper on the development of a guideline on the demonstration of therapeutic equivalence for locally applied and locally acting products in the gastrointestinal tract, EMA/CHMP/558326/2013, www.ema.europa.eu.
The note for guidance on the clinical requirements for locally applied, locally acting products containing known constituents provides general recommendations on the clinical requirements for respective formulations with known active substances. This concept paper discusses the need to expand the guidance on locally applied and locally acting gastrointestinal products.
Draft: consultation open, Consultation end date 31/12/2013.
- European Medicines Agency, Concept paper on the development of a guideline on the demonstration of therapeutic equivalence for locally applied and locally acting products in the gastrointestinal tract, EMA/CHMP/558326/2013, www.ema.europa.eu.
The note for guidance on the clinical requirements for locally applied, locally acting products containing known constituents provides general recommendations on the clinical requirements for respective formulations with known active substances. This concept paper discusses the need to expand the guidance on locally applied and locally acting gastrointestinal products.
Draft: consultation open, Consultation end date 31/12/2013.
- European Medicines Agency, Draft reflection paper on the data requirements for intravenous iron-based nano-colloidal products developed with reference to an innovator medicinal product, EMA/CHMP/SWP/620008/2012, www.ema.europa.eu.
This reflection paper discusses the data requirements for nano-sized colloidal intravenous iron-based preparations developed as a treatment for iron deficiency with reference to a nano-sized colloidal innovator products.(...)
Draft: consultation open, Consultation end date 28/02/2014. - European Medicines Agency, Draft guideline on the clinical development of medicinal products for the treatment of human-immunodeficiency-virus (HIV) infection (revision 3), EMEA/CPMP/EWP/633/02 Rev 3, www.ema.europa.eu.
This guideline provides guidance on the clinical development of direct-acting antiretrovirals for the treatment of human-immunodeficiency-virus (HIV) infection. It replaces EMEA/CPMP/EWP/633/02 Rev 2.
Draft: consultation open, Consultation end date 31/03/2014. - European Medicines Agency, Draft paediatric addendum to the note for guidance on the clinical investigation on medical products in treatment of hypertension, EMA/CHMP/206815/2013, www.ema.europa.eu.
This is an addendum to the guideline on clinical investigation of medicinal products in the treatment of hypertension (EMA/238/1995/Rev. 3, 18 November 2010). It is not meant as a guidance document on its own but rather highlights differences from adult patients with arterial hypertension and points out paediatric-specific aspects.
Draft: consultation open, Consultation end date 31/03/2014. - European Medicines Agency, Draft guideline on the clinical development of medicinal products for the treatment of human-immunodeficiency-virus (HIV) infection (revision 3), EMEA/CPMP/EWP/633/02; Rev 3, www.ema.europa.eu.
This guideline provides guidance on the clinical development of direct-acting antiretrovirals for the treatment of human-immunodeficiency-virus (HIV) infection. It replaces EMEA/CPMP/EWP/633/02 Rev 2.
Draft: consultation open, Consultation end date 31/03/2014.
Zonder dienstverleningsovereenkomst niet voldaan aan de eisen van onderzoek
CGR Codemissie Advies, 5 september 2013 AA13.064 Negatief advies, medisch wetenschappelijk onderzoek. Namens [vergunninghouder Z] heeft verzoekster gevraagd een verklaring van geen bezwaar af te geven voor het uitvoeren van het onderzoek met de titel “[A]”. [A] is een retrospectieve analyse van patiëntencasussen met een [aandoening B]-geschiedenis in meerdere landen. De primaire doelstellingen van het onderzoek zijn het vaststellen van baseline patiëntenkenmerken, het type [aandoening B] en de gerelateerde ziekenhuisprocedures, en de behandelpatronen (in het bijzonder het gebruik van [geneesmiddelen C]) tijdens een meer dan 18 maanden durende post-[aandoening B] follow up periode.
Negatief advies, medisch wetenschappelijk onderzoek. Namens [vergunninghouder Z] heeft verzoekster gevraagd een verklaring van geen bezwaar af te geven voor het uitvoeren van het onderzoek met de titel “[A]”. [A] is een retrospectieve analyse van patiëntencasussen met een [aandoening B]-geschiedenis in meerdere landen. De primaire doelstellingen van het onderzoek zijn het vaststellen van baseline patiëntenkenmerken, het type [aandoening B] en de gerelateerde ziekenhuisprocedures, en de behandelpatronen (in het bijzonder het gebruik van [geneesmiddelen C]) tijdens een meer dan 18 maanden durende post-[aandoening B] follow up periode.
De beoordeling door de Codecommissie
(...)
Op grond van het bovenstaande dient in de eerste plaats te worden nagegaan of het onderzoek als niet-WMO-plichtig kan worden aangemerkt. De WMO is van toepassing op onderzoek dat aan twee voorwaarden voldoet, te weten: er is sprake van medisch wetenschappelijk onderzoek en de proefpersonen worden aan handelingen onderworpen en/of aan proefpersonen wordt een bepaalde gedragswijze opgelegd. Hiervan is in dit geval geen sprake. Het onderzoek zal bestaan uit een multinationale, niet-interventionele, retrospectieve analyse van casussen van een nader aangegeven aantal patiënten. Er kan daarom inderdaad van niet-WMO-plichtig onderzoek gesproken worden.
Vervolgens moet worden nagegaan of is voldaan aan de eisen voor een dergelijk onderzoek. In de Nadere Uitwerking, die hierboven al genoemd is, wordt als eerste eis gesteld dat de dienstverleningsovereenkomst met inbegrip van de te verrichten diensten en tegenprestaties schriftelijk moet zijn vastgelegd. Daarbij is voorts vermeld dat deze eis niet geldt voor overeenkomsten die enkel strekken tot het eenmalig invullen van eenvoudige vragenlijsten dan wel enquête-formulieren. Deze eis stelt de vraag aan de orde of in het onderhavige geval een dienstverleningsovereenkomst vereist is en zo ja, of daarin is voorzien.
Naar het oordeel van de Codecommissie kan niet worden aangenomen dat de uitzonderingsbepaling toepassing zou kunnen vinden. De vragen die door de deelnemende artsen beantwoord zullen moeten worden, zijn omvangrijk en kunnen niet als eenvoudig worden aangemerkt. Bovendien gaat het niet om een eenmalig invullen van een vragenlijst, immers zullen de deelnemende artsen over het algemeen meerdere vragenlijsten invullen. Er is immers vastgelegd dat een deelnemende arts maximaal 10 patiënten mag includeren.Bij de stukken die door verzoekster zijn overgelegd, bevindt zich geen dienstverleningsovereenkomst. De afspraken die met de deelnemende artsen moeten worden gemaakt, komen op een andere manier tot stand. De artsen zullen in de eerste plaats lid moeten worden van [organisatie D]. Dat brengt mee dat zij accoord gaan met de algemene voorwaarden tussen artsen en [D]. Daarna kunnen de artsen uitgenodigd worden om deel te nemen aan een onderzoek. De voorwaaren van dat onderzoek worden via e-mail aan de arts bekend gemaakt. [D] nodigt de artsen die zij willen laten deelnemen uit met een brief waarin de taken omschreven zijn en welk bedrag de artsen daarvoor zullen ontvangen. De artsen kunnen vervolgens op een link klikken en aangeven dat zij aan het onderzoek willen deelnemen.
De Codecommissie is van oordeel dat deze gang van zaken onvoldoende duidelijkheid geeft. Van verzoekster moet worden verlangd dat zij in één stuk de deelnemende artsen uiteenzet welke prestaties van hen verwacht worden en welke prestaties daar dan van de andere kant tegenover worden gesteld. Voorts moet worden verlangd dat zij dit alles in een schriftelijk
stuk vast legt. In dit verband verwijst de Codecommissie naar artikel 12 van de Uitwerking Normen Gunstbetoon artikelen 12 en 13, 16 t/m 22 Gedragscode Geneesmiddelenreclame waar deze laatste eis met zo veel woorden in is opgenomen.
Nu naar het oordeel van de Codecommissie aan de eerste eis die in deze gesteld is, niet wordt voldaan, ziet de Codecommissie geen aanleiding op de verder gestelde eisen in te gaan. Volledigheidshalve worden die eisen alleen kort aangegeven: de doelstelling en uitvoering van het onderzoek dienen helder te zijn omschreven, de doelstelling moet zinvol en legitiem zijn en de opzet en uitvoering dienen voldoende kwaliteit te waarborgen.
Een en ander leidt tot de conclusie dat een negatief advies moet worden gegeven. De Codecommissie gaat er daarbij van uit dat verzoekster een advies heeft bedoeld te vragen. Een verklaring van geen bezwaar kent de regelgeving waar de Codecommissie zich aan houdt, niet.
Uitvoer van wetenschappelijk onderzoek naar H5N1-virus is vergunningplichtig
Rechtbank Noord-Holland 20 september 2013, ECLI:NL:RBNHO:2013:8527 (X tegen Minister voor Buitenlandse Handel en Ontwikkelingssamenwerking)
 Persbericht Rechtspraak.nl: De rechtbank Noord-Holland heeft op 20 september 2013 geoordeeld dat de uitvoer van (wetenschappelijke) manuscripten die onderzoeksresultaten bevatten over H5N1-virustechnologie vergunningplichtig is. Het H5N1-virus is een variant van het vogelpestvirus dat gevaarlijk is voor mensen.
Persbericht Rechtspraak.nl: De rechtbank Noord-Holland heeft op 20 september 2013 geoordeeld dat de uitvoer van (wetenschappelijke) manuscripten die onderzoeksresultaten bevatten over H5N1-virustechnologie vergunningplichtig is. Het H5N1-virus is een variant van het vogelpestvirus dat gevaarlijk is voor mensen.
In het kader van Verordening (EG) nr. 428/2009 moeten de lidstaten (waaronder Nederland) van de Europese Unie een adequaat en doeltreffend controlesysteem inrichten ter voorkoming van de verspreiding van onder meer biologische wapens. Hoewel de Verordening zelf geen uitzonderingen kent, is in de bijlage bij de Verordening bepaald dat de vergunningsregelingen voor overdracht van “technologie” niet van toepassing zijn op informatie die “voor iedereen beschikbaar” is en op “fundamenteel wetenschappelijk onderzoek”.
Uitzonderingsgronden
Belanghebbende beroept zich op deze uitzonderingsgronden en meent dat er voor de uitvoer van deze manuscripten daarom geen vergunningplicht geldt. De Minister voor Buitenlandse Handel en Ontwikkelingssamenwerking is van oordeel dat er wel een vergunning nodig is, die in dit geval overigens ook is verleend. De rechtbank stelt de Minister dus in het gelijk.
Uitzonderingen op de vergunningplicht moeten strikt worden uitgelegd
Gelet op het doel van de Verordening, het voorkomen van verspreiding van materialen die kunnen bijdragen tot de ontwikkeling en verspreiding van biologische wapens, moeten de uitzonderingen op de vergunningplicht strikt worden uitgelegd. Deze beperkte uitleg brengt mee dat de uitzondering op de vergunningplicht voor “fundamenteel wetenschappelijk onderzoek” slechts geldt voor onderzoek dat niet is gericht op het realiseren van een praktisch doel in verband met de verspreiding van biologische wapens. Eiser heeft met het onderzoek aangetoond dat het mogelijk is om het desbetreffende virus via de lucht overdraagbaar te maken. Dit is volgens de rechtbank een praktisch doel en zou daarom zonder vergunningplicht het doel van de Verordening in gevaar brengen. Het is verder niet zo dat de onderzoekers alleen methoden hebben gebruikt die al beschikbaar waren en eerder zijn gepubliceerd. De onderzoekers hebben namelijk stappen gezet en keuzes gemaakt die tot geheel nieuwe uitkomsten hebben geleid. Namelijk dat het mogelijk is om het desbetreffende virus via de lucht overdraagbaar te maken.
Uitsluiting bevoegde autoriteiten
Tegen de uitspraak van de rechtbank staat hoger beroep open bij het gerechtshof.
Willekeurig een alternatief kiezen van een analyse, is niet inventief
Rechtbank Den Haag 2 oktober 2013, ECLI:NL:RBDHA:2013:15067 (TEVA Pharma tegen Sanofi) Octrooirecht. ABC. Zie ook: IEF 13018. Sanofi is houder van Aanvullend Beschermingscertificaat (het combinatie-ABC) voor irbesartan, desgewenst in de vorm van een zout en/of een hydraat, en hydrochloorthiazide (HTCTZ). Irbesartan en HCTZ worden gebruikt voor de behandeling van hoge bloeddruk, een veel voorkomende aandoening die kan leiden tot ernstige vaatziekten. Dit combinatie-ABC is verleend op basis van een een EP 0454 511. Teva vordert vernietiging van het combinatie-ABC, met veroordeling van Sanofi in de proceskosten ex 1019h Rv.
Octrooirecht. ABC. Zie ook: IEF 13018. Sanofi is houder van Aanvullend Beschermingscertificaat (het combinatie-ABC) voor irbesartan, desgewenst in de vorm van een zout en/of een hydraat, en hydrochloorthiazide (HTCTZ). Irbesartan en HCTZ worden gebruikt voor de behandeling van hoge bloeddruk, een veel voorkomende aandoening die kan leiden tot ernstige vaatziekten. Dit combinatie-ABC is verleend op basis van een een EP 0454 511. Teva vordert vernietiging van het combinatie-ABC, met veroordeling van Sanofi in de proceskosten ex 1019h Rv.
De rechtbank beoordeelt de inventiviteit van het onderliggende Europees octrooi. De rechtbank kijkt naar de meest nabij gelegen stand van techniek, een artikel van Chiu en een octrooi van Du Pont. Sanofi heeft willekeurig één van de alternatieven, beschikbaar na de analyse van Du Pont, gekozen en dat is niet inventief. Daarmee valt de grondslag voor het combinatie ABC weg en moet het ABC op basis van artikel 15 van de ABC-verordening worden vernietigd. Sanofi wordt als de in het ongelijk gestelde partij in de proceskosten veroordeeld.
De beoordeling
4.16. De rechtbank overweegt als volgt. Op zich is juist, zoals Sanofi stelt, dat in de regel een “pointer” of motivatie nodig is om de gemiddelde vakman de richting naar de uitvinding te wijzen wil inventiviteit ontbreken. Ten onrechte echter trekt Sanofi daaruit de conclusie dat die “pointer” of motivatie bij het enkel vinden van een alternatief voor de dichtstbijzijnde stand van de techniek, specifiek en eenduidig naar (uitsluitend) de geclaimde oplossing dient te wijzen. Alle alternatieven moeten immers als een oplossing van het gestelde probleem worden gezien, zolang van die alternatieven redelijkerwijs kan worden verwacht dat ze werkzaamheid behouden, een gemiddelde vakman deze in overweging zou nemen en hij ze op conventionele wijze kan synthetiseren zonder “undue burden”. Dat betekent evenzeer dat alle alternatieven die aan die voorwaarden voldoen, niet inventief zijn. Zie de volgende uitspraken van de Technische Kamers van beroep van het EOB: T0821/97, r.o. 6.4; T0852/91, r.o. 8.2; T0892/08, r.o. 1.7; T0964/92, r.o. 2.10, T0879/05, r.o. 5.3, T0631/06, r.o. 2.3.10.4.17. Anders gezegd, bij gebreke aan een reden (zoals een onverwacht, verbeterd effect, hetgeen bij irbesartan niet aan de orde is zoals hiervoor onder 4.5. en 4.6. overwogen), is een keuze van één alternatief dat naar verwachting werkzaamheid behoudt uit vele (die na SAR-analyse van DuP 753 beschikbaar zijn) willekeurig en aldus niet inventief te achten. Zie een lange lijn van uitspraken van de Technische Kamers van beroep van het EOB: T0892/08, r.o. 1.7; T0964/92, r.o. 2.10, T0879/05, r.o. 5.3, T0631/06, r.o. 2.3.10, T0345/07, r.o. 4.7.3, T0012/07, r.o. 4,1,6, T0423/08, r.o. 2.3.5, T0931/04, r.o. 4.11.1.
4.18. Aldus kan er geen andere conclusie zijn dan dat conclusie 7 niet inventief is. Dat Du Pont irbesartan niet ook heeft gevonden en geclaimd in haar octrooiaanvragen maakt dit oordeel niet anders. Sanofi heeft de gemotiveerde stelling van Teva dat alsdan ook conclusie 20, irbesartan in combinatie met een diureticum, voor de hand ligt, niet bestreden zodat ook die conclusie moet sneuvelen. Evenmin heeft Sanofi betwist dat bij die stand van zaken de grondslag voor het combinatie ABC is weggevallen en het ABC op basis van artikel 15 van de ABC-verordening moet worden vernietigd. De overige argumenten van Teva behoeven gelet hierop geen nadere bespreking meer.
Lees de uitspraak hier:
Rechtspraak (link)
Rechtspraak (pdf)
HA ZA 12-1209 (pdf)
CBP: Advies over uitbreiding verstrekking medische gegevens
Uit het persbericht: CBP adviseert over uitbreiding verstrekking medische gegevens geestelijke gezondheidszorg aan zorgverzekeraars, cbpweb.nl 2 oktober 2013.jpg) Wetgevingsadvies. Het College bescherming persoonsgegevens (CBP) heeft advies uitgebracht over het aan zorgverzekeraars verstrekken van informatie over de zorgvraagzwaarte in de curatieve geestelijke gezondheidszorg (ggz). Hulpverleners zouden op declaraties de zogeheten zorgvraagzwaarte-indicator moeten vermelden. Met deze regeling wordt beoogd tegemoet te komen aan knelpunten die zorgverzekeraars ervaren bij de wettelijk verplichte controle van declaraties. Het CBP heeft bezwaren tegen de regeling in de voorgestelde vorm en adviseert deze niet aldus in te voeren.
Wetgevingsadvies. Het College bescherming persoonsgegevens (CBP) heeft advies uitgebracht over het aan zorgverzekeraars verstrekken van informatie over de zorgvraagzwaarte in de curatieve geestelijke gezondheidszorg (ggz). Hulpverleners zouden op declaraties de zogeheten zorgvraagzwaarte-indicator moeten vermelden. Met deze regeling wordt beoogd tegemoet te komen aan knelpunten die zorgverzekeraars ervaren bij de wettelijk verplichte controle van declaraties. Het CBP heeft bezwaren tegen de regeling in de voorgestelde vorm en adviseert deze niet aldus in te voeren.
Privacygevoelige medische gegevens
Door de voorgestelde wijziging van de Regeling zorgverzekering (Rzv) wordt het aantal persoonsgegevens uitgebreid dat hulpverleners in de ggz aan zorgverzekeraars moeten doorgeven en waarvoor zij het medisch beroepsgeheim moeten doorbreken. Deze hulpverleners zouden voortaan niet alleen verplicht zijn de diagnosebehandelcombinatie (DBC; gegevens over de diagnose van een patiënt) aan zorgverzekeraars te verstrekken, maar ook de zorgvraagzwaarte-indicator. Dit is een getal dat een indicatie geeft van de aard en omvang van de hulpverlening.
In beide gevallen betreft het medische persoonsgegevens die zeer privacygevoelig zijn. Met name in de ggz waar deze gegevens de kern van het privéleven van de betrokken persoon raken moet uiterst zorgvuldig en terughoudend worden omgegaan met patiëntgegevens en met doorgifte hiervan aan derden die niet bij de behandeling betrokken zijn. De noodzaak, proportionaliteit en subsidiariteit van de voorgestelde regeling dienen daarom zorgvuldig onderbouwd en afgewogen te worden.
Advies CBP
Het CBP adviseert om de verplichte verstrekking van informatie over de zorgvraagzwaarte in de curatieve ggz te beperken tot die diagnosegroepen waarbij het aannemelijk is dat de zorgvraagzwaarte-indicator ook daadwerkelijk voorspellende waarde heeft voor de behandelinzet. Uit onderzoek is namelijk gebleken dat dit niet altijd het geval is.
Verder adviseert het CBP om de effecten van invoering van de zorgvraagzwaarte-indicator zorgvuldig te monitoren. Mede op basis daarvan kan worden besloten over bredere invoering en/of verdere ontwikkeling van de zorgvraagzwaarte-indicator of een ander geschikt instrument.
Tot slot adviseert het CBP om twee uitzonderingen mogelijk te maken op de verplichting om de zorgvraagzwaarte-indicator te verstrekken, zoals dat ook bij vermelding van de DBC geregeld is. De eerste uitzondering zou moeten gelden als de rekening direct door de verzekerde zelf wordt betaald en dus niet ten laste wordt gebracht van de zorgverzekering, de tweede als patiënt en hulpverlener een zogeheten privacyverklaring ondertekenen.
HvJ EU: Richtlijn medische hulpmiddelen staat indeling product als geneesmiddel toe
HvJ EU 3 oktober 2013, zaak C-109-/12 (Labaratoires Lyocentre) - dossier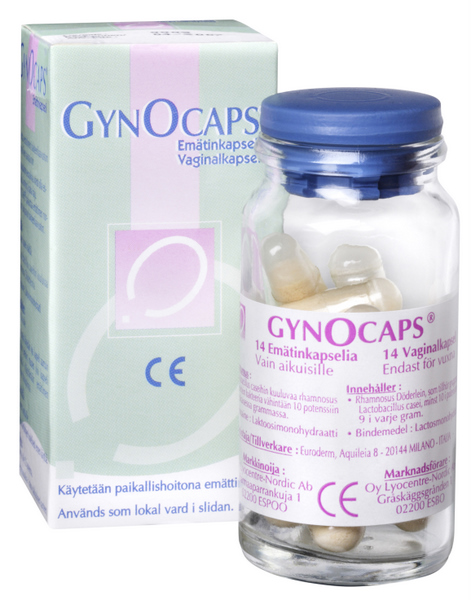 Zie eerder LSenR. Verzoek om een prejudiciële beslissing, Korkein hallinto-oikeus. Uitlegging van richtlijn 93/42/EEG van de Raad van 14 juni 1993 betreffende medische hulpmiddelen (PB L 169, blz. 1) en richtlijn 2001/83/EG van het Europees Parlement en de Raad van 6 november 2001 tot vaststelling van een communautair wetboek betreffende geneesmiddelen voor menselijk gebruik (...). Vaginaal preparaat met levende melkzuurbacteriën. Recht van bevoegde nationale instantie om een in een andere lidstaat als medisch hulpmiddel verkocht preparaat met een EG-markering in de zin van richtlijn 93/42 op basis van zijn farmacologisch, immunologisch of metabolisch effect te kwalificeren als geneesmiddel in de zin van richtlijn 2001/83. Het Hof verklaart voor recht:
Zie eerder LSenR. Verzoek om een prejudiciële beslissing, Korkein hallinto-oikeus. Uitlegging van richtlijn 93/42/EEG van de Raad van 14 juni 1993 betreffende medische hulpmiddelen (PB L 169, blz. 1) en richtlijn 2001/83/EG van het Europees Parlement en de Raad van 6 november 2001 tot vaststelling van een communautair wetboek betreffende geneesmiddelen voor menselijk gebruik (...). Vaginaal preparaat met levende melkzuurbacteriën. Recht van bevoegde nationale instantie om een in een andere lidstaat als medisch hulpmiddel verkocht preparaat met een EG-markering in de zin van richtlijn 93/42 op basis van zijn farmacologisch, immunologisch of metabolisch effect te kwalificeren als geneesmiddel in de zin van richtlijn 2001/83. Het Hof verklaart voor recht:
1) Het feit dat een product in een lidstaat krachtens richtlijn 93/42/EEG van de Raad van 14 juni 1993 betreffende medische hulpmiddelen, (...) als medisch hulpmiddel met EG-markering is ingedeeld, staat niet eraan in de weg dat de bevoegde autoriteiten van een andere lidstaat ditzelfde product wegens het farmacologische, immunologische of metabolische effect ervan indelen als geneesmiddel in de zin van artikel 1, punt 2, sub b, van richtlijn 2001/83/EG van het Europees Parlement en de Raad van 6 november 2001 tot vaststelling van een communautair wetboek betreffende geneesmiddelen voor menselijk gebruik (...).
2) De bevoegde autoriteiten van een lidstaat moeten, teneinde een product dat in een andere lidstaat krachtens richtlijn 93/42, zoals gewijzigd bij richtlijn 2007/47, reeds is ingedeeld als medisch hulpmiddel met EG-markering, krachtens richtlijn 2001/83, zoals gewijzigd bij verordening nr. 1901/2006, in te delen als geneesmiddel, de procedure van artikel 18 van richtlijn 93/42, zoals gewijzigd bij richtlijn 2007/47, en, in voorkomend geval, deze van artikel 8 van die richtlijn 93/42 toepassen alvorens zij de indelingsprocedure van richtlijn 2001/83, zoals gewijzigd bij verordening nr. 1901/2006, toepassen.
3) Binnen eenzelfde lidstaat kan een product dat weliswaar niet identiek is aan een ander, als geneesmiddel ingedeeld product, maar daarmee toch eenzelfde bestanddeel gemeen heeft en hetzelfde werkingsmechanisme heeft, in beginsel niet krachtens richtlijn 93/42, zoals gewijzigd bij richtlijn 2007/47, in de handel worden gebracht als medisch hulpmiddel, tenzij een ander uit het oogpunt van artikel 1, lid 2, sub a, van die richtlijn 93/42 relevant kenmerk dat eigen is aan een dergelijk product vereist dat dit product als medisch hulpmiddel wordt aangemerkt en in de handel wordt gebracht, wat door de verwijzende rechter dient te worden beoordeeld.
Gestelde vragen:
1) Wanneer een preparaat in een lidstaat overeenkomstig richtlijn 93/42 is gekwalificeerd als een medisch hulpmiddel met een EG-markering in de zin van deze richtlijn, is het dan uitgesloten dat de bevoegde autoriteit van een andere lidstaat dit preparaat op basis van het farmacologische, immunologische of metabolische effect ervan indeelt als geneesmiddel in de zin van artikel 1, lid 2, sub b, van richtlijn [2001/83]?
2) Indien de vorige vraag ontkennend wordt beantwoord, kan deze nationale autoriteit het preparaat alleen met inachtneming van de procedures van richtlijn [2001/83] als geneesmiddel indelen of moeten, alvorens de procedure tot indeling als geneesmiddel volgens deze richtlijn wordt begonnen, de vrijwaringsprocedure van artikel 8 van richtlijn [93/42] of de voorschriften over onrechtmatig aangebrachte EG-markeringen in de zin van artikel 18 van deze richtlijn worden gevolgd?
3) Sluiten richtlijn [2001/83], richtlijn [93/42] of andere bepalingen van Unierecht (met name inzake de bescherming van de gezondheid en het leven van de mens en de consumentenbescherming) uit dat preparaten met hetzelfde bestanddeel en dezelfde werkingsmechanismen op het grondgebied van één en dezelfde lidstaat op de markt zijn als zowel geneesmiddel dat een vergunning voor het in de handel brengen in de zin van richtlijn 2001/83/EG vereist, als medisch hulpmiddel in de zin van richtlijn [93/42]?
Nieuw niche kantoor Van Innis en Delarue
 Former Allen & Overy colleagues Thierry van Innis en Dieter Delarue launch new Belgian IP boutique law firm, with offices in Antwerp and Brussels: Van Innis & Delarue.
Former Allen & Overy colleagues Thierry van Innis en Dieter Delarue launch new Belgian IP boutique law firm, with offices in Antwerp and Brussels: Van Innis & Delarue.
Thierry van Innis and Dieter Delarue announce the formation of VAN INNIS & DELARUE, a boutique law firm for creative and innovative clients, focusing on intellectual property law and related subject matters. Many existing clients are trend setting in media & entertainment, designer goods, life sciences and e-commerce & technology.
VAN INNIS & DELARUE brings together more than forty years of experience at leading international law firms. The work of the firm’s lawyers in intellectual property matters before the national courts, the Benelux Court of Justice and the Court of Justice of the European Union has led to dozens of precedents and landmark decisions. Thierry van Innis, referred to by Chambers and Partners as “the pope of trade mark law in Belgium” and qualified as a “Senior Statesman” for IP law in Belgium, has litigated more high profile trade mark cases before the Court of Justice of the European Union and the Benelux Court of Justice than any other IP lawyer. He is also still the only Belgian ever who has won a Lifetime Achievement Award at the World Leaders European IP Awards.Referring to the rationale behind the plans, Thierry van Innis commented:
“Many of our creative and innovative clients have worked with IP boutique law firms abroad, and praise the specialized IP boutique as a law firm model where lawyers can be as passionate and innovative as they are about their brands, creations and inventions. Also, they deliver the right mix of experience and specialization on one hand, and availability and a personal touch on the other hand”.
“While the specialized boutique is a law firm model which is commonly used in Belgium for other specialist areas such as tax and employment law, it is relatively new for IP matters”, Dieter Delarue added. “We are committed to position our boutique law firm among the handful of ‘go-to’ IP litigation firms in the Belgian market. Our mission is to provide clients with the highest possible quality of service. In this respect, we are pleased to begin working with a small group of people who know each other, who have worked together before, and who are all dedicated to excellence in advocacy”.
Thierry van Innis and Dieter Delarue re-unite in this new boutique, having previously worked together at Allen & Overy, until Thierry’s move to Field Fisher Waterhouse in 2009. The team will be strengthened by the arrival of Heidi Waem, an associate who also joins from Allen & Overy. The firm’s plan calls for measured growth over the next two to three years, with the objective to build a focused and specialized team of four to eight attorneys.
For more information and extensive CVs of the founders, please visit www.vaninnis-delarue.be.
Omzetbelasting heffen bij alternatieve geneeskundigen
Rechtbank Den Haag 2 oktober 2013, ECLI:NL:RBDHA:2013:12925 (AVIG en NAAV) Niet-ontvankelijk. Eigen belang AVIG is een beroepsvereniging van artsen voor homeopathie, natuurgeneeskunde, biofysische geneeskunde en neuraaltherapie. NAAV is een beroepsvereniging van artsen en tandartsen voor accupuntuur. Op 1 januari 2013 is de wet van 12 juli 2012 tot wijziging van onder meer de Wet Omzetbelasting 1968 in werking getreden. Hierin staat BTW-uitsluiting voor gezondheidskundige diensten, dit is uitgewerkt door de staatssecretaris van Financiën. De uitsluiting geldt niet voor artsen die ook alternatieve en/of complementaire behandelmethoden aanbieden.
Niet-ontvankelijk. Eigen belang AVIG is een beroepsvereniging van artsen voor homeopathie, natuurgeneeskunde, biofysische geneeskunde en neuraaltherapie. NAAV is een beroepsvereniging van artsen en tandartsen voor accupuntuur. Op 1 januari 2013 is de wet van 12 juli 2012 tot wijziging van onder meer de Wet Omzetbelasting 1968 in werking getreden. Hierin staat BTW-uitsluiting voor gezondheidskundige diensten, dit is uitgewerkt door de staatssecretaris van Financiën. De uitsluiting geldt niet voor artsen die ook alternatieve en/of complementaire behandelmethoden aanbieden.
AVIG en NAAV worden niet-ontvankelijk verklaard. Zij hadden geen eigen belang te behartigen in deze procedure. Het gaat om de belangen van de leden. Op grond van de Algemene wet inzake Rijksbelastingen hadden ze bezwaar moeten maken en vervolgens in beroep gaan bij de bestuursrechter. Toetsing door de bestuursrechter kan pas nadat daadwerkelijk uitvoerig door middel van belastingheffing plaatsvindt. De voorzieningenrechter kijkt niet inhoudelijk naar het geschil.
De beoordeling
4.3 Kern van het geschil betreft de vraag of omzetbelasting mag worden geheven bij de leden van AVIG en NAAV voor de door hen geleverde diensten. Met het oog op het gesloten stelsel van rechtsmiddelen is de bestuursrechter bij uitsluiting bevoegd om te oordelen over de rechtmatigheid van de op aangifte van individuen te betalen belasting. Uit artikel 26 lid 2 van de Algemene wet inzake Rijksbelastingen vloeit immers voort dat tegen de voldoening van omzetbelasting bezwaar kan worden gemaakt en vervolgens beroep bij de bestuursrechter kan worden ingesteld. In die procedure kunnen alle bezwaren die in deze procedure zijn aangevoerd aan de orde worden gesteld. Anders dan AVIG en NAAV c.s. kennelijk betogen, lijdt het geen twijfel dat de rechter in die procedure bevoegd en zelfs verplicht is de stelling te beoordelen dat sprake is van strijdigheid met Europese regelgeving voor zover dat nodig is voor de beslissing op het door die rechter behandelde beroep. Voorts bepaalt artikel 8:81 lid 1 van de Algemene wet bestuursrecht dat de (bestuursrechtelijke) voorzieningenrechter, zodra bezwaar is gemaakt (op verzoek) een voorlopige voorziening kan treffen in geval van onverwijlde spoed. Een en ander brengt mee dat door de wetgever een speciale rechtsgang is aangewezen die met voldoende waarborgen is omkleed, ook indien zich een spoedeisend geval voordoet. Daar komt bij dat een behoorlijke taakverdeling tussen de burgerlijke rechter en de bestuursrechter het in het algemeen ongewenst doet zijn dat tegelijkertijd voor beide rechters procedures over de verbindendheid van voorschriften worden gevoerd, met het risico van een verschillende uitkomst. [eiser 1] c.s. dienen dan ook niet-ontvankelijk te worden verklaard in hun vorderingen.
4.4. De omstandigheid dat (nieuwe) regelgeving niet op voorhand door de bestuursrechter kan worden getoetst, maar eerst nadat daadwerkelijk uitvoering door middel van belastingheffing plaatsvindt, maakt het voorgaande niet anders. Die omstandigheid vloeit immers voort uit het gesloten stelsel van rechtsbescherming in het belastingrecht. Deze door de wetgever gewilde beperking levert niet een rechtstekort op waarin door de burgerlijke rechter zou moeten worden voorzien (HR 21 april 2006, NJ 2006, 271). De in dit arrest bedoelde uitzondering waarin door de burgerlijke rechter kan worden getoetst of een voorgenomen besluit of standpunt van de inspecteur onmiskenbaar onjuist of zonder grond is, doet zich in deze zaak niet voor nu reeds bezwaar is en kan worden gemaakt tegen de voldoening van de omzetbelasting en het handelen van de (mogelijk) belastingplichtige niet meer door het standpunt van de inspecteur zal worden beïnvloed. Bovendien hebben [eiser 1] c.s. deze kortgedingprocedure pas enkele maanden na de inwerkingtreding van de nieuwe regelgeving aanhangig gemaakt, zodat toetsing vóór de inwerkingtreding hoe dan ook niet meer aan de orde is. Dat [eiser 1] c.s. zelf handelingen moeten verrichten die tot daadwerkelijke uitvoering van de regelgeving en daarmee tot het openen van een rechtsingang bij de bestuursrechter leiden, leidt evenmin tot een ander oordeel. [eiser 1] c.s. zijn immers verplicht tot het verrichten van die handelingen (het doen van aangifte) en zijn daartoe kennelijk – gelet op de mededeling dat reeds bezwaarprocedures lopen – ook al overgegaan.
4.5 Kern van het geschil betreft de vraag of omzetbelasting mag worden geheven bij de leden van AVIG en NAAV voor de door hen geleverde diensten. Met het oog op het gesloten stelsel van rechtsmiddelen is de bestuursrechter bij uitsluiting bevoegd om te oordelen over de rechtmatigheid van de op aangifte van individuen te betalen belasting. Uit artikel 26 lid 2 van de Algemene wet inzake Rijksbelastingen vloeit immers voort dat tegen de voldoening van omzetbelasting bezwaar kan worden gemaakt en vervolgens beroep bij de bestuursrechter kan worden ingesteld. In die procedure kunnen alle bezwaren die in deze procedure zijn aangevoerd aan de orde worden gesteld. Anders dan AVIG en NAAV c.s. kennelijk betogen, lijdt het geen twijfel dat de rechter in die procedure bevoegd en zelfs verplicht is de stelling te beoordelen dat sprake is van strijdigheid met Europese regelgeving voor zover dat nodig is voor de beslissing op het door die rechter behandelde beroep. Voorts bepaalt artikel 8:81 lid 1 van de Algemene wet bestuursrecht dat de (bestuursrechtelijke) voorzieningenrechter, zodra bezwaar is gemaakt (op verzoek) een voorlopige voorziening kan treffen in geval van onverwijlde spoed. Een en ander brengt mee dat door de wetgever een speciale rechtsgang is aangewezen die met voldoende waarborgen is omkleed, ook indien zich een spoedeisend geval voordoet. Daar komt bij dat een behoorlijke taakverdeling tussen de burgerlijke rechter en de bestuursrechter het in het algemeen ongewenst doet zijn dat tegelijkertijd voor beide rechters procedures over de verbindendheid van voorschriften worden gevoerd, met het risico van een verschillende uitkomst. [eiser 1] c.s. dienen dan ook niet-ontvankelijk te worden verklaard in hun vorderingen.
4.4. De omstandigheid dat (nieuwe) regelgeving niet op voorhand door de bestuursrechter kan worden getoetst, maar eerst nadat daadwerkelijk uitvoering door middel van belastingheffing plaatsvindt, maakt het voorgaande niet anders. Die omstandigheid vloeit immers voort uit het gesloten stelsel van rechtsbescherming in het belastingrecht. Deze door de wetgever gewilde beperking levert niet een rechtstekort op waarin door de burgerlijke rechter zou moeten worden voorzien (HR 21 april 2006, NJ 2006, 271). De in dit arrest bedoelde uitzondering waarin door de burgerlijke rechter kan worden getoetst of een voorgenomen besluit of standpunt van de inspecteur onmiskenbaar onjuist of zonder grond is, doet zich in deze zaak niet voor nu reeds bezwaar is en kan worden gemaakt tegen de voldoening van de omzetbelasting en het handelen van de (mogelijk) belastingplichtige niet meer door het standpunt van de inspecteur zal worden beïnvloed. Bovendien hebben [eiser 1] c.s. deze kortgedingprocedure pas enkele maanden na de inwerkingtreding van de nieuwe regelgeving aanhangig gemaakt, zodat toetsing vóór de inwerkingtreding hoe dan ook niet meer aan de orde is. Dat [eiser 1] c.s. zelf handelingen moeten verrichten die tot daadwerkelijke uitvoering van de regelgeving en daarmee tot het openen van een rechtsingang bij de bestuursrechter leiden, leidt evenmin tot een ander oordeel. [eiser 1] c.s. zijn immers verplicht tot het verrichten van die handelingen (het doen van aangifte) en zijn daartoe kennelijk – gelet op de mededeling dat reeds bezwaarprocedures lopen – ook al overgegaan.
MvT: Wijziging van de Tabakswet ter invoering van een verhoogd strafmaximum op overtreding van het rookverbod
Memorie van Toelichting, Wijziging van de Tabakswet ter invoering van een verhoogd strafmaximum op overtredig van het rookverbod (Verhoging strafmaximum overtredinig rookverbod Tabakswet), Kamerstukken II, 2013/14, 33738 nr. 3 1. Inleiding - De huidige Tabakswet kent twee wettelijke strafmaxima voor wat betreft op te leggen bestuurlijke boeten:
1. Inleiding - De huidige Tabakswet kent twee wettelijke strafmaxima voor wat betreft op te leggen bestuurlijke boeten:
a. € 450 000 voor overtreding van het reclameverbod door fabrikanten, groothandels of importeurs van tabaksproducten;
b. € 4.500 voor alle overige overtredingen.
De regering acht het gewenst deze twee trappen aan te vullen met een tussenliggend maximum van € 19 500 voor het overtreden van het rookverbod.(...)
4. Gevolgen voor burgers, bedrijfsleven, overheid en milieu - Het wetsvoorstel heeft geen gevolgen voor overheid en milieu. Het wetsvoorstel als zodanig heeft evenmin directe gevolgen voor burgers en bedrijfsleven, terwijl het ook geen (nieuwe) administratieve lasten voor burgers of bedrijfsleven met zich brengt. Uiteraard wordt met dit wetsvoorstel wel de weg geopend om in de toekomst overtredingen van het rookverbod bestuurlijk zwaarder te beboeten dan thans het geval is en daar kunnen de overtredende burgers en bedrijven uiteraard wel de gevolgen van ondervinden.
Voor volledige tekst: Kamerstukken II, 2013/14, 33738 nr. 3
Landelijke eHealth-monitor 2013
Bijlage bij Kamerstukken II 2013/2014, 27529, nr. 126. (Landelijke eHealth-monitor 2013)%20wikimedia.png) Nederland staat voor de uitdaging om gezondheidszorg van goede kwaliteit, toegankelijk en betaalbaar te houden. Vergrijzing leidt tot meer behoefte aan zorg, terwijl tegelijkertijd het aantal mensen dat zorg kan verlenen afneemt. Inmiddels heeft een kwart van de Nederlanders een of meer chronische ziekten zoals COPD en hart- en vaatziekten (RIVM, 2013). Een groot deel van de zorgkosten betreft zorg aan deze groep. Om de stijging van uitgaven in de zorg af te remmen en om noodzakelijke zorg met de beschikbare menskracht te kunnen blijven bieden, is het nodig zorg anders te organiseren.
Nederland staat voor de uitdaging om gezondheidszorg van goede kwaliteit, toegankelijk en betaalbaar te houden. Vergrijzing leidt tot meer behoefte aan zorg, terwijl tegelijkertijd het aantal mensen dat zorg kan verlenen afneemt. Inmiddels heeft een kwart van de Nederlanders een of meer chronische ziekten zoals COPD en hart- en vaatziekten (RIVM, 2013). Een groot deel van de zorgkosten betreft zorg aan deze groep. Om de stijging van uitgaven in de zorg af te remmen en om noodzakelijke zorg met de beschikbare menskracht te kunnen blijven bieden, is het nodig zorg anders te organiseren.














